Formas de amiloidosis cardíaca y presentación clínica
Se presentó una actualización sobre la amiloidosis cardíaca, enfermedad de depósito caracterizada por la acumulación de proteína mal plegada en forma de fibrillas amiloides. Las dos formas principales que afectan al corazón son la amiloidosis por cadenas ligeras (AL) y la amiloidosis por transtiretina (TTR), esta última en sus variantes wild type (senil) y mutada (hereditaria). La forma AL es una enfermedad multisistémica con manifestaciones características como fragilidad capilar, hematomas perioculares y macroglosia, requiriendo derivación a hematología para tratamiento específico. Sin tratamiento, el pronóstico vital es infausto en pocos meses.
La amiloidosis por transtiretina en su forma senil afecta principalmente al corazón, presentándose como miocardiopatía restrictiva, mientras que la forma hereditaria asocia mayor afectación del sistema nervioso periférico con polineuropatía y disautonomía. En Baleares existe el quinto foco mundial y primer foco español de la forma hereditaria, con más de 150 casos controlados, mayoritariamente por la mutación Val50Met. Aunque tradicionalmente considerada enfermedad rara en su forma hereditaria, la variante senil no lo es: hasta el 16% de pacientes con miocardiopatía hipertrófica, 16% con estenosis aórtica mayor de 65 años y 18% con insuficiencia cardíaca con fracción de eyección preservada tienen amiloidosis cardíaca subyacente.
Los red flags clínicos incluyen: insuficiencia cardíaca con NT-proBNP desproporcionadamente elevado, trastornos de conducción, síndrome del túnel del carpo bilateral, estenosis de canal lumbar, polineuropatía, disautonomía y antecedentes familiares. El perfil típico del paciente con forma wild type es varón mayor de 65 años (>80% varones) con túnel del carpo y supervivencia sin tratamiento de 2-6 años. La forma mutada afecta a pacientes de 30-60 años, con menor predominio masculino, polineuropatía marcada y supervivencia variable (algunos casos de solo 3 años). Las mujeres representan un desafío diagnóstico: menor frecuencia, peor supervivencia, menos hipertrofia ventricular y diagnóstico más tardío.
Algoritmo diagnóstico y herramientas clave
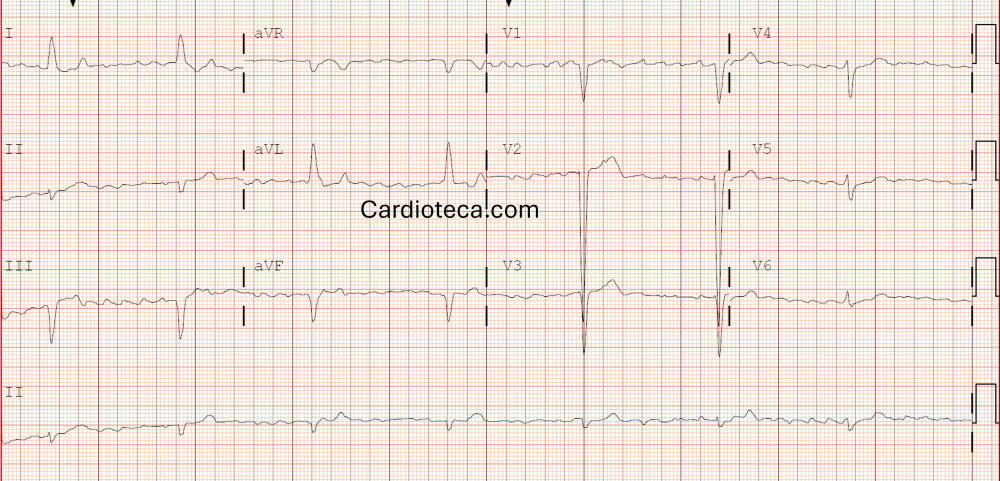
Se enfatizó que el diagnóstico requiere alta sospecha clínica. El algoritmo diagnóstico comienza con historia clínica detallada, exploración física, electrocardiograma, ecocardiograma y resonancia cardíaca (preferiblemente con T1 mapping y volumen extracelular, que permiten detectar enfermedad en estadios precoces). El electrocardiograma mostró ser fundamental: aunque clásicamente se describían bajos voltajes, lo más frecuente es el patrón de pseudoinfarto anterior, trastornos de conducción y fibrilación auricular (especialmente en la forma senil).
Una vez establecida la sospecha, el segundo escalón diagnóstico incluye gammagrafía cardíaca y estudio hematológico simultáneo (cuantificación de cadenas ligeras en suero, inmunofijación en suero y orina de 24 horas). Es crucial realizar ambas pruebas en paralelo, ya que hasta el 25% de gammagrafías positivas (captación estadio 2-3 de Perugini) corresponden a amiloidosis AL. Si la gammagrafía es positiva y el estudio hematológico negativo, se procede al estudio genético para diferenciar entre forma wild type o mutada. El ecocardiograma con strain longitudinal global mostrando el patrón "cherry on top" (respeto apical) es muy característico de miocardiopatías infiltrativas.
La simplificación diagnóstica con gammagrafía y resonancia ha evitado biopsias endomiocárdicas y ha reducido el retraso diagnóstico, históricamente de 3-4 años. Se recomendó screening activo en pacientes con hipertrofia ventricular izquierda ≥12 mm más uno o más red flags. Se destacaron proyectos de búsqueda proactiva: detección de captaciones incidentales en gammagrafías oncológicas (100% confirmadas como amiloidosis), biopsias de túnel del carpo y estenosis de canal lumbar (8% de amiloidosis cardíaca detectada), y estudios de genómica, microbiota y proteómica en desarrollo. La calculadora TAMY facilita el diagnóstico, con score ≥7 puntos indicando alta probabilidad de amiloidosis.
Tratamiento actual y perspectivas futuras
El tratamiento se divide en soporte y específico. El manejo de soporte incluye anticoagulación directa en fibrilación auricular (sin calcular CHA₂DS₂-VASc, dado el 30% de riesgo de trombos), precaución extrema con digoxina (prácticamente contraindicada), uso cauteloso de betabloqueantes, y preferencia por antialdosterónicos y diuréticos de asa en insuficiencia cardíaca. Los IECA/ARA-II no aportan beneficio.
El tratamiento farmacológico específico ha revolucionado el pronóstico. Los estabilizadores de transtiretina incluyen tafamidis (una dosis diaria, reducción de mortalidad demostrada en estudio ATTR-ACT, NNT 7-8 pacientes para prevenir una muerte a 30 meses) y acoramidis (dos dosis diarias, potencia del 96% vs 50% del tafamidis, también con reducción de mortalidad demostrada). Los silenciadores de ARN (patisirán, vutrisirán, eplontersen) reducen la producción hepática de transtiretina; vutrisirán ya demostró beneficios cardiovasculares en New England Journal of Medicine y próximamente se aprobará para cardiopatía además de polineuropatía.
El futuro inmediato incluye edición génica mediante CRISPR con una única infusión (ensayo MAGNITUDE fase III en marcha) y deflexionadores que eliminan depósitos ya acumulados de amiloide (fase I-II muy alentadores). Se concluyó que la amiloidosis cardíaca ya no es sentencia de muerte ni discapacidad: el diagnóstico precoz y los tratamientos disponibles cambian radicalmente la supervivencia y calidad de vida. La supervivencia a 60 meses de cohortes actuales versus históricas muestra diferencias impresionantes. El mensaje principal fue la necesidad de búsqueda proactiva y sospecha sistemática en escenarios clínicos de riesgo.

Elena Fortuny Frau